Children born with propionic acidemia may have delays in development. This condition is treated by dietary restriction. The most common symptom of this condition is attacks of ketoacidosis - a situation where fat is used for energy instead of glucose.
Propionic acidemia is a rare disorder that is not specific to any ethnic group, sex, or population group. Many people who are born with this condition appear normal at birth but start to develop symptoms that include: small size at birth, abnormally developed muscle tone, poor feeding, lethargy, consentient vomiting, dehydration, ketoacidosis (build up of ketones in the blood and can be detected by an abnormal smell in the mouth) and seizures.
Causes of Propionic Acidemia
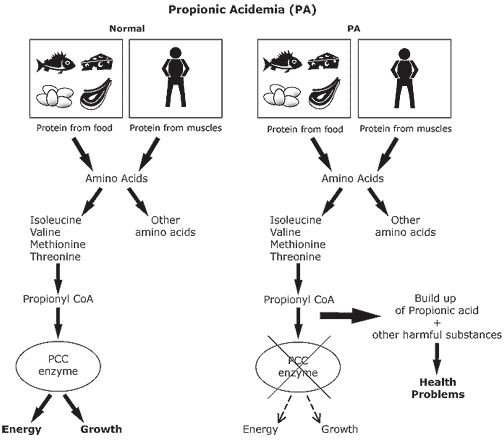
Propionic acidemia is caused by mutations in the PCCA or PCCB genes. The PCCA and PCCB genes encode for the enzyme propionyl-CoA carboxylase (PCC), which is the specific enzyme that is either missing or not working properly. When the PCC enzyme is not working properly, the body is not able to break down proteins and fats properly. This leads to a buildup of the amino acid glycine and a fatty acid called propionic acid in the blood stream. As a result of the accumulated propionic acid in the blood, damage can occur to the lining of blood vessels, leading to the possibility that the propionic acid can leak into the brain and nerve tissues. This can lead to serious health problems, such as seizures, developmental delay, and problems with muscle strength (weak ability to walk and talk) (Desviat 2006, Ugarte 1999).
Patients that have a mutation in the PCCA gene are put into a complementation group pccA and patients with a mutation in the PCCB gene are put into a complementation group called pccBC. Patients that are in the pccA group tend to have a more severe form of the disorder because the PCC enzyme deficiency does more harm to the body. Since the PCCA gene encodes for an alpha subunit and the PCCAB gene encodes from a beta subunit (two parts of the PCC enzyme), the location of the mutation in the PCCA and PCCB gene and whether a person inherits one or two abnormal copies of either gene will ultimately determine the severity of the condition(Desviat 2006, Ugarte 1999).
A Basic Overview of Propionic Acidemia
How Propionic Acidemia is Treated
Propionic acidemia is treated by the use of a protein supplement that does not contain the four amino acids (isoleucine, valine, threonine, and methionine) that are precursors for the formation of the enzyme propionyl-CoA carboxylase. The protein supplement that is used, which is a cow milk formula, still allows for formation of propionate, which is needed for normal health. Families need to consult with a biochemical geneticist and nutritionist to develop a dietary plan for restricting the intake of the four amino acids to only the amounts required for normal growth, as the four amino acids are essential dietary needs (Fenton 2001, Ney 1985).
The four amino acids, isoleucine, valine, threonine, and methionine, are responsible for toxicity (presence of ketone bodies) being present in the blood when consumed in amounts greater than needed for normal growth (Childs 1961).
Prenatal Diagnosis
Prenatal diagnosis can be done by the use of chorionic villus (CV) sampling (Muro 1999).
References
Childs B, Hyhan WL, Borden MA, et al. (1961). Idiopathic hyperglycinemia and hyperglycinuria, a new disorder of amino acid metabolism. Pediatrics 27:522.
Desviat, L. R.; Clavero, S.; Perez-Cerda, C.; Navarrete, R.; Ugarte, M.; Perez, B. (2006). New splicing mutations in propionic acidemia. Journal of Human Genetics 51: 992-997.
Fenton, W. A.; Gravel, R. A.; Rosenblatt, D. S. Disorders of propionate and methylmalonate metabolism. In: Scriver, C. R.; Beaudet, A. L.; Sly, W. S.; Valle, D. (eds.) : The Metabolic and Molecular Bases of Inherited Disease. Vol. II. (8th ed.) New York: McGraw-Hill 2001. P. 2176.
Muro, S.; Perez-Cerda, C.; Rodriguez-Pombo, P.; Perez, B.; Briones, P.; Ribes, A.; Ugarte, M. (1999). Feasibility of DNA based methods for prenatal diagnosis and carrier detection of propionic acidaemia. Journal of Medical Genetics 36: 412-414.
Ney DN, Bay C, Saudubray JM, et al. (1985). An evaluation of protein requirements in methylmaloic acidaemia. Journal of Inherited Metabolic Disease 8:132.
Ugarte M, Perez-Cerda C, Rodriquez-Pombo P, Desviat LR, Perez B, Richard E, Muro S, Campeau E, Ohura T, Gravel RA. (1999). Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Human Mutation 14(4):275-82.


