Apert syndrome is a rare genetic disorder involving malformations of the skull, face, hands and feet. It is caused by mutations on chromosome 10. Find out more about what causes Apert syndrome, its symptoms, and how it is inherited.
What is Apert Syndrome?
According to The National Institutes of Health (2008) Apert syndrome is a rare genetic disorder whose main condition is craniosynostosis, that is the premature fusion of skull bones, affecting, thus the face and head of the patient. Also, and as a consequence of the disease, another condition known as syndactyly, is manifested. In syndactyly, fingers and toes are fused together.
What Causes Apert Syndrome?
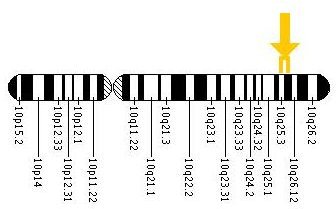
Apert syndrome is a genetic disease (or genetic defect) caused by a mutated gene. More specifically, it is caused by mutations in the fibroblast growth factor receptor 2 gene (FGFR2 ), located on chromosome 10. The FGFR2 protein is involved in embryonic development, cell division and growth, and cell maturation. The figure bellow (HIH, 2008, public domain) shows the exact location of the gene mutations that causes Apert syndrome.
In regard to Apert syndrome, almost all cases are caused by any of two mutations in the FGFR2 gene. These are simple mutations in which one of the amino acids of the proteins is changed. In one mutation a serine amino acid (located in position 252) is changed by a tryptophan (mutation Ser252Trp). The second mutation is a Pro253Arg in which a proline amino acid is replaced by an arginine amino acid at position 253. These single point mutations cause the FGFRG2 protein to lose its proper functioning and consequently the symptoms of Apert disease.
Location of Mutations on Chromosome 10 (NIH, 2008, Public domain)
How is Apert Syndrome Inherited?
Apert syndrome is an autosomal dominant type of disease. This means that only one copy of the altered gene is needed for the disease to occur. Accordingly, an offspring of a parent with Apert syndrome has a 50% chance of getting the mutations and thus the disease.
The Apert condition may also result from spontaneous mutations of the genetic material of newborns whose parents have no history of the disorder in their family. The occurrence of the disease is estimated at 1 in 65,000 to 88,000 newborns.
Apert Syndrome Treatments
There is no cure for the Apert syndrome genetic defects, but there is much that can be done to manage the symptoms of the disease, including surgery to separate fused bones (most patients will require several operations), and speech therapy to help overcome difficulties with talking that are encountered by some people.
Sources:
National Institutes of Health (2008). Apert syndrome. Genetics Home Reference (available at https://ghr.nlm.nih.gov/condition=apertsyndrome (accessed on September 8th, 2008).
Wilkie et al (1995). Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome". Nature genetics 9: 165–72.


