De novo design of proteins starts with generating a protein with a specific sequence. The sequence is engineered to result in a complex, folded, functional protein. This field poses challenges that are being overcome rapidly as the technology advances.
Background: Protein Structure
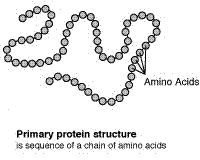
Proteins are composed of chains of molecules called amino acids. All known life uses only twenty amino acids, all with sinistral (left-handed) symmetry, and their arrangement results in the astonishing diversity of proteins that occur in nature. The amino acid chains are called peptides (especially long ones are called polypeptides), and they fold due to chemical interactions among constituent atoms. The resulting molecules can be highly complex, with up to four levels of structure.
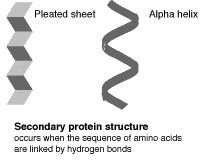
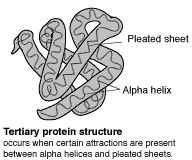
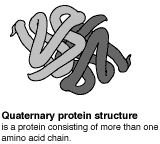
The primary (level 1) structure of a protein is the sequence of amino acids in a polypeptide. The amino acids are linked through peptide bonds, and DNA codes directly for their sequence. Hydrogen bonds among the amino acids can shape a polypeptide into a regular coil (alpha helix) or pleat (beta sheet), resulting in secondary (level 2) structure. Folding into a tertiary (level 3) structure results from interactions among alpha helices and beta sheets, such as ionic bonds (salt bridges), sulfide bonds, and hydrogen bonds. Many proteins consist of two or more folded polypeptides (subunits) connected through covalent or sulfide bonds, and this is the fourth (quaternary) level of protein structure.
The Four Levels of Protein Structure
Reverse Engineering of Protein Structure
De novo protein design requires designing a primary structure that leads to the desired tertiary level of structure. Understanding how primary structure leads to secondary and tertiary structure begins by deconstructing existing proteins. The first step is to decode the primary structure through protein sequencing.
Short peptides of no more than about 50 amino acids can be sequenced using a technique called the Edman degradation. Longer polypeptides can be sequenced through mass spectrometry; John Bennett Fenn won the 2002 Nobel Prize in chemistry for developing a good method using this technique. Amino acid sequence can also be predicted, rather than determined empirically, by sequencing and decoding the DNA or RNA that encodes it in the genome.
A real protein molecule may contain thousands of amino acids, all of which interact to result in three or four levels of structure. Real proteins are so complex that they cannot be modeled “as-is,” atom by atom, with current computing technology (though DNA computers may one day make this more feasible). Instead, they are simplified in two ways. First, instead of representing every atom, the simulation represents each amino acid as a whole, like a bead with specific properties. Second, the beads are modeled in a rigid cubical lattice, from which comes the name of these representations: lattice proteins.
About a decade ago, this technology allowed de novo protein design to become a reality. “De novo” is a Latin phrase literally meaning “from the new,” and de novo protein design is the engineering of proteins from scratch. The first de novo designed proteins were reported near the end of the 20th century (for example, Dahiyat & Mayo 1997).
Problems in the Energy Phases of De Novo Proteins
Proteins are known to have at least two distinct phases. The “native state” is the energetic ground state and also the state at which the protein functions biologically. It has a stable tertiary structure. The denatured state is the “unfolded” state of the protein and has no tertiary structure. Another phase of proteins, the molten globule, has a dynamic, unstable tertiary structure. The molten globule may be a transitional state or a third distinct thermodynamic state (Pande & Rokhsar 1998).
A challenge in de novo protein design has been achieving a stable native state; researchers have been frustrated by results that remain in the molten globule state. Progress is being made, with sequence-designed proteins with stable tertiary structure having been reported (for example, Offreti et al. 2003).
The future promises great progress in the de novo engineering of designer proteins. This area of the field of biotechnology holds great promise.


