The Killer Karyotype of Patau Syndrome: What's So Unlucky About Trisomy 13?
Nature, for all its wonder and beauty, can be a cold and unforgiving mother. The machinery of evolution, after all, depends as much on the reality of death as it does on regenerating new life: if old life did not give way to new, there would be no capacity for any species to evolve. This is the darker aspect of what we call survival of the fittest, wherein nature acts not only to reward those organisms with genes coding for advantageous traits, but also to strike down the unlucky organism whose genetic material varies too widely from the time-tested parameters of the status quo. Genetic variability is certainly the sine qua non of evolutionary change, but nature will tolerate only so much of it from one generation to the next. A variation too drastic is met with severe and quite final consequences for any such wayward would-be progeny. For all mammals, including humans, this results in spontaneous abortion of the fetus which, if it occurs late enough in pregnancy, yields what we would additionally recognize as a stillbirth.
Research indicates that approximately half of all spontaneous abortions in humans are found to have an abnormal chromosomal complement, meaning a deviation from the usual number of 46 chromosomes per cell. To review, a normal human cell contains 23 pairs of chromosomes, with one set of 23 originating from the individual’s mother, and the other set from the father; the only exception to this rule are the reproductive cells, or gametes (i.e., egg or sperm), which contain only half of a complete set, thus allowing a newly conceived zygote to inherit the normal complement of 46 chromosomes when egg and sperm fuse at the moment of conception. Of these, one pair, the sex chromosomes, will ultimately determine the fetus’ gender– the familiar XY or XX combination, which yield a male or female, respectively. The remaining 22 pairs of chromosomes are called autosomes, meaning simply that they do not code for gender-related characteristics. An autosomal trisomy, then, occurs when an embryo winds up having three copies of a given non-sex chromosome instead of the usual two. The mechanism by which this can happen varies, but most often it is the result of a non-disjunction error occurring during maternal oogenesis, or egg production. Such an error results in an egg cell that has 24 instead of the normal 23 chromosomes, so that when conception occurs and the sperm introduces its set of 23 chromosomes into the newly formed zygote, the total number is now 47 instead of 46. Note that similar errors can and do occur during spermatogenesis as well, and there is a small percentage of cases of trisomy attributable to paternal origins, but for the vast majority of cases the opposite holds true.
As we’ve already suggested, the outcome for the resulting embryo/fetus is not good. In most cases spontaneous abortion occurs either prior to implantation or at some point along the course of development, resulting in miscarriage or stillbirth. However, it turns out that some trisomies are more lethal than others. For example, trisomy 16 (where the embryo inherits three copies of chromosome #16 from its parents instead of two) is the autosomal trisomy found most commonly in spontaneously aborted fetuses, and no such fetus has ever progressed to a live birth. In contrast, trisomy 21, better known as Down syndrome, is the most common trisomy among live births, resulting in a fetus and, for the roughly 25% of cases that do not expire in utero, a newborn with many characteristic abnormalities, including mental retardation and certain congenital heart defects, but one that is nonetheless frequently viable well into adulthood, albeit with both a level of functioning and a life expectancy markedly reduced from that of the general population. For any fetus conceived with an autosomal trisomy, this is essentially the best-case scenario. Of all the other potential trisomies, only trisomy 18 (Edwards syndrome) and trisomy 13 (Patau syndrome) are known to result in a viable fetus with any significant degree of regularity, with the rest either having never resulted in a live birth or having done so in only a handful of reported cases over the past several hundred years.
As might be expected, the clinical outcome in either Edwards or Patau syndrome is less than ideal. Though the specifics are outside the scope of this article, suffice it to say that the congenital defects resulting from either trisomy are typically more profound than those seen in Down syndrome, with a much higher mortality rate. Nevertheless, a minority of fetuses with trisomy 18 or trisomy 13 do survive into infancy and beyond. For the infant with Patau syndrome, while the diagnosis is often clear in the presence of the characteristic cluster of birth defects, a karyotype analysis is the only way to definitively confirm the occurrence of trisomy 13. As mentioned, the most common cause of trisomy 13 is a non-disjunction error during the first meiotic phase of oogenesis, meaning there is a failure of separation of the mother’s 13th pair of chromosomes, ultimately resulting in an egg cell with one too many copies of chromosome 13. If this egg is ever fertilized by a sperm, the resulting zygote will consequently contain 47 chromosomes instead of 46, with three copies of chromosome 13 instead of two.
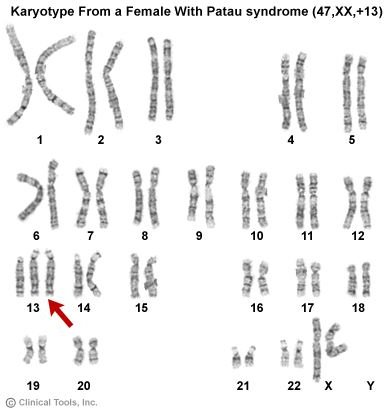
The resulting karyotype for this individual would appear as follows:
The notation for this karyotype, 47,XX,+13, signifies that there are 47 chromosomes, the two sex chromosomes are XX (i.e., the individual is female), and the additional chromosome is an extra copy of chromosome 13.
While a non-disjunction error is the underlying cause for most cases of trisomy 13, in a minority of cases the extra copy of chromosome 13 arises from what is termed a Robertsonian translocation. In this case, the karyotype reveals not three copies of chromosome 13 but the usual two copies; however, elsewhere in the karyotype an additional copy of the long arm of chromosome 13 is essentially fused onto the top of another chromosome. Typically, this translocation involves chromosome 14, as shown in the following example:
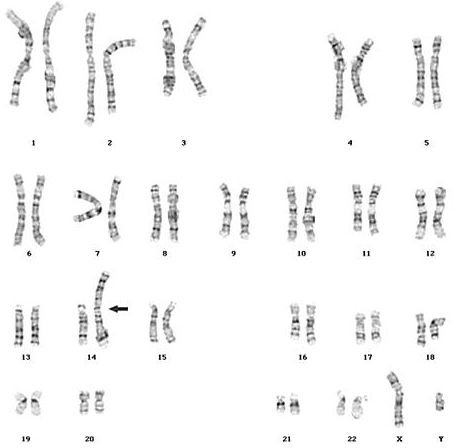
Here, the karyotype notation would be 46,XY,+13,dic(13;14), signifying that there are 46 chromosomes, the sex chromosomes are XY (i.e., the individual is male), there is an extra copy of chromosome 13, which appears as a dicentric chromosome comprising both chromosomes 13 and 14. Note that since the additional copy of chromosome 13 is structurally part of a larger single chromosome that also includes the long arm of chromosome 14, technically there are still only 46 chromosomes in the karyotype. Functionally, however, the clinical outcome is the same as would be seen in the more common non-disjunction error derived situation, and the affected individual suffers all the same consequences that go along with full-blown Patau syndrome.
There is a milder form of Patau syndrome that occurs in what is termed “mosaic” trisomy 13, where only some of the body’s cells contain the extra copy of chromosome 13, while the rest have a normal chromosomal complement. In order for this to happen, there needs to have been a non-disjunction or translocation error that occurs not during the formation of the parents’ reproductive cells, but instead during embryonic mitosis, the process of cell division by which the developing embryo’s cells multiply themselves to produce the trillions of cells ultimately comprising the fully-developed fetus. Since only a subset of the individual’s cells contain the chromosomal aberration, he or she is spared the often devastating consequences of a “full” trisomy 13. The degree of mildness or severity depends on the proportion of cells carrying the trisomy, which is in turn a function of how early in embryogenesis the mitotic error occurred: the earlier the error, the greater the proportion of subsequent cells that contain it, and the more severely abnormal the resulting phenotype. The karyotype in the case of a mosaic trisomy refers to each distinct population of cells (i.e., normal and aberrant), and varies somewhat depending on the specific case in question. For example, for a male individual in which a mitotic non-disjunction error occurred at some point during embryogenesis, the karyotype would be denoted 47,XY,+13/46,XY.
References
Jones, KL. Smith’s Recognizable Patterns of Human Malformation, 6th ed, Elsevier Saunders, Philadelphia 2006.
Tolmie, JL. Down syndrome and other autosomal trisomies. In: Emery and Rimoin’s Principles and Practice of Medical Genetics, 3rd ed, Rimoin, DL, Connor, JM, Pyeritz, RE, Churchill Livingston 1996. p.925.
Warburton, D, Kline, J, Stein, Z, et al. Cytogenetic abnormalities in spontaneous abortions of recognized conception. In: Perinatal Genetics Diagnosis and Treatment, Porter, IH, Willey, A (Eds), Academic Press, New York 1986. p.133.
Image credits
Karyotype for Trisomy 13 (NDE-type): obtained from Clinical Tools, Inc. and used under Creative Commons License.
Karyotype for Trisomy 13 (RT-type): courtesy of the Seattle, WA Dynacare Cytogenetics Laboratory.