Glaucoma, an eye disease affecting millions and the leading cause of irreversible blindness worldwide, is not a single disease entity, but a common outcome of a wide variety of ocular pathology, some genetic in basis, some not. Here we explore what is known about heritable forms of glaucoma.
Overview
Glaucoma refers to a group of eye diseases characterized by progressive degeneration of the optic nerve, resulting first in peripheral vision loss, followed by total, irreversible blindness if left untreated. Unfortunately, it is quite common, second only to cataracts in terms of the number of cases of blindness it causes across the globe . Though there is an unmistakable correlation between glaucoma and elevated intraocular pressure (IOP), meaning a higher than normal internal pressure of the aqueous fluid filling the eye, the relationship between elevated IOP and optic nerve damage is not clear-cut: some people who have higher than normal IOP may never go on to develop glaucoma, whereas others with severe glaucoma are found to have IOP entirely within the “normal” range. As such, though it is fairly certain that elevations in IOP play some manner of causative role in the etiology of glaucomatous optic nerve damage, the mechanism by which this occurs is not completely understood.
Nevertheless, as with every disease process, understanding its genetic underpinnings helps us better understand how and why it occurs, and can guide us to newer, safer and more effective treatments. However, because many different diseases affecting the eye can result in glaucoma, either alone or as part of a larger syndrome of ocular problems, its inheritance is not straightforward. Though the picture is indeed rather murky, there are a number of identified genes that confer an increased risk of glaucoma to carriers.
Inside the Eye
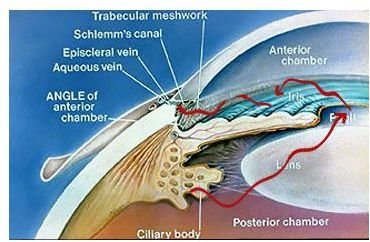
Before we touch upon those it will be helpful first to briefly review the relevant anatomy of the eye. The eye is essentially a hollow ball filled with clear fluid, with a hole (the pupil) on one side to let light in, and on the opposite side, a specialized lining (the retina) capable of converting incoming light into electrochemical impulses. A bundle of nerve cells (the optic nerve) emerges from the retina and transmits these electrochemical impulses back to the visual centers of the brain, where they can then be consciously experienced and interpreted in the phenomenon we call sight. In the anterior chamber of the eye (
), which contains the lens, iris, pupil and cornea, a porous structure called the trabecular meshwork exists in the angle formed at the junction of the iris (the colored part of the eye surrounding the pupil) and cornea (a translucent bubble of tissue that covers the iris and helps focus incoming light through the pupil and lens). This is the angle in question when glaucoma is classified as open or closed angle . The trabecular meshwork acts as a kind of filter through which the aqueous humor circulates on its way out of the eyeball’s interior into the veins located on its surface (the sclera, or “white” of the eye) to be drained away. If the outflow of this fluid is obstructed in some way, inadequate drainage will result in increased intraocular pressure, placing the eye at greatly increased risk of developing glaucoma.
Primary vs. Secondary Glaucoma
In the broadest terms, glaucoma can be divided into two categories: primary glaucoma, wherein a congenital abnormality of an isolated structure in the anterior chamber directly impairs aqueous outflow, and secondary glaucoma, wherein outflow obstruction is either acquired through external means (e.g., trauma, infection, etc.) or occurs as part of a more pervasive underlying disease process. There are three main types of primary glaucoma: primary open angle glaucoma (POAG), juvenile open angle glaucoma (JOAG) and primary congenital glaucoma (PCG), each with a unique pattern of inheritance. Though many of the causes of secondary glaucoma are heritable disorders, none of them consistently result in glaucoma in all cases and so the genetics of these disorders will not be discussed here (even patients with Sturge-Weber syndrome , classically characterized as a triad of unilateral glaucoma, facial port-wine stain and intracranial angiomata, will never develop glaucoma in as many as 70% of cases). To keep things simple, we will focus instead on how the three main types of primary glaucoma are inherited.
Primary Open Angle Glaucoma
POAG is the most common of the primary glaucomas, affecting over 33 million people worldwide. It is a disease of old age, with people age 65 and older accounting for over 99% of the affected population. There is a gender disparity as well, as it affects women at approximately twice the rate it does men. Race is another significant risk factor, with studies showing a prevalence of POAG in the African-American population exceeding that of Caucasian-Americans by almost sevenfold. The most current research suggests that POAG is a phenotype resulting from the interaction of multiple genes. Its mode of inheritance is thus not straightforward, though overall we see a pattern approximating autsomal dominant transmission with incomplete penetrance. Thus far, three genes have been identified as conferring an increased risk of developing POAG, named MYOC, OPTN and WDR36.
MYOC, formerly known as TIGR (trabecular-meshwork induced glucocorticoid response gene), is located on the long arm of chromosome 1 and codes for myocilin, a protein whose function has not been fully characterized. It has been found floating in solution in the aqueous humor of the eye, but its areas of highest concentration are the iris, sclera and trabecular meshwork. It is also found in the myelin sheath of the optic as well as peripheral nerves. Over seventy point mutations in the MYOC gene have been identified across multiple ethnic groups. An interplay among specific mutations in MYOC with mutations in other genes has been observed. For example, a Gly399Val mutation in MYOC results in the POAG phenotype, with a mean age of onset of 51 years; however, when this mutation occurs in the presence of an Arg368His mutation in the CYP1B1 gene (a PCG-associated gene, as we will see shortly), the mean age of onset decreases markedly, to 27 years. Though not entirely understood, the consensus is that defects in the myocilin protein result in some kind of trabecular meshwork dysfunction, which in turn contributes to increased IOP and finally glaucoma.
OPTN codes for optineurin, a protein of uncertain function expressed in the trabecular meshwork, retina and non-pigmented ciliary epithelium of the eye. It is associated with less than 1% of cases of POAG, but is found with greater relative frequency in the subgroup of glaucoma sufferers having “normal” levels of IOP. Regarding WDR36, there is conflicting evidence in support of its playing a causative role in POAG, though it seems clear that it does have some modulating effect on the severity of the glaucoma that develops in the presence of the other genes. It codes for a protein expressed in a number of intraocular structures that influences T-cell activation and proliferation, suggesting a potential autoimmune component to the pathophysiology of POAG.
References
Causes of Blindness and Visual Impairment, WHO website, https://www.who.int/blindness/causes/en/ , accessed on 2/1/2010.
Challa, P. Glaucoma genetics. International Ophthalmology Clinics, 2004; 44(2):167-185.
Enjolras, O, Riche, MC, Merland, JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics 1985; 76:48.
Kipp, MA. Childhood glaucoma. Pediatric Clinics of North America, 2003; 50:89.
Image Credits
Anterior Chamber of the Eye: courtesy of Davidson Eye Associates, PA


